publications
A pdf version of the full list of publications can be downloaded here: list of publications.
2026
- Angular-dependent interatomic potential for large-scale simulation of bcc and hcp multi-component refractory alloysSergei Starikov, Petr Grigorev, Sang-Hyeok Lee, Zhuocheng Xie, and Pär A T OlssonComputational Materials Science 262 p. 114369 (Jan 2026)
This work is devoted to the development and comprehensive validation of a new interatomic potential for bcc and hcp refractory alloys based on the WMoNbTaZrTi system. The presented model allows the simulation of various structural transformations, as well as the behavior of crystal defects in several of the phases observed in this system. The classical form of the potential enables simulations of atomic systems comprising up to 108 atoms for durations longer than a million time steps using a routine computational setting. The wide applicability of the developed model is demonstrated by the example of studying phase transformations in TiNb alloys and the properties of defects in Laves phases.
2025
- A foundation model for atomistic materials chemistryIlyes Batatia, Philipp Benner, Yuan Chiang, Alin M Elena, Dávid P Kovács, Janosh Riebesell, Xavier R Advincula, Mark Asta, Matthew Avaylon, William J Baldwin, Fabian Berger, Noam Bernstein, Arghya Bhowmik, Filippo Bigi, Samuel M Blau, Vlad Cărare, Michele Ceriotti, Sanggyu Chong, James P Darby, Sandip De, Flaviano Della Pia, Volker L Deringer, Rokas Elijošius, Zakariya El-Machachi, Edvin Fako, Fabio Falcioni, Andrea C Ferrari, John L A Gardner, Mikołaj J Gawkowski, Annalena Genreith-Schriever, Janine George, Rhys E A Goodall, Jonas Grandel, Clare P Grey, Petr Grigorev, Shuang Han, Will Handley, Hendrik H Heenen, Kersti Hermansson, Cheuk Hin Ho, Stephan Hofmann, Christian Holm, Jad Jaafar, Konstantin S Jakob, Hyunwook Jung, Venkat Kapil, Aaron D Kaplan, Nima Karimitari, James R Kermode, Panagiotis Kourtis, Namu Kroupa, Jolla Kullgren, Matthew C Kuner, Domantas Kuryla, Guoda Liepuoniute, Chen Lin, Johannes T Margraf, Ioan-Bogdan Magdău, Angelos Michaelides, J Harry Moore, Aakash A Naik, Samuel P Niblett, Sam Walton Norwood, Niamh O’Neill, Christoph Ortner, Kristin A Persson, Karsten Reuter, Andrew S Rosen, Louise A M Rosset, Lars L Schaaf, Christoph Schran, Benjamin X Shi, Eric Sivonxay, Tamás K Stenczel, Christopher Sutton, Viktor Svahn, Thomas D Swinburne, Jules Tilly, Cas Oord, Santiago Vargas, Eszter Varga-Umbrich, Tejs Vegge, Martin Vondrák, Yangshuai Wang, William C Witt, Thomas Wolf, Fabian Zills, and Gábor CsányiThe Journal of Chemical Physics 163 p. 184110 (Nov 2025)
Atomistic simulations of matter, especially those that leverage first-principles (ab-initio) electronic structure theory, provide a microscopic view of the world, underpinning much of our understanding of chemistry and materials science. Over the last decade or so, machine-learned force fields have transformed atomistic modeling by enabling simulations of ab-initio quality over unprecedented time and length scales. However, early machine-learning (ML) force fields have largely been limited by (i) the substantial computational and human effort required to develop and validate potentials for each particular system of interest and (ii) a general lack of transferability from one chemical system to the next. Here, we show that it is possible to create a general-purpose atomistic ML model, trained on a public dataset of moderate size, that is capable of running stable molecular dynamics for a wide range of molecules and materials. We demonstrate the power of the MACE-MP-0 model—and its qualitative and at times quantitative accuracy—on a diverse set of problems in the physical sciences, including properties of solids, liquids, gases, chemical reactions, interfaces, and even the dynamics of a small protein. The model can be applied out of the box as a starting or ’foundation’ model for any atomistic system of interest and, when desired, can be fine-tuned on just a handful of application-specific data points to reach ab-initio accuracy. Establishing that a stable force-field model can cover almost all materials changes atomistic modeling in a fundamental way: experienced users obtain reliable results much faster, and beginners face a lower barrier to entry. Foundation models thus represent a step toward democratizing the revolution in atomic-scale modeling that has been brought about by ML force fields.
- Exploring parameter dependence of atomic minima with implicit differentiationIvan Maliyov, Petr Grigorev, and Thomas D. Swinburnenpj Computational Materials 11 p. 22 (Jan 2025)
Interatomic potentials are essential to go beyond ab initio size limitations, but simulation results depend sensitively on potential parameters. Forward propagation of parameter variation is key for uncertainty quantification, whilst backpropagation has found application for emerging inverse problems such as fine-tuning or targeted design. Here, the implicit derivative of functions defined as a fixed point is used to Taylor-expand the energy and structure of atomic minima in potential parameters, evaluating terms via automatic differentiation, dense linear algebra or a sparse operator approach. The latter allows efficient forward and backpropagation through relaxed structures of arbitrarily large systems. The implicit expansion accurately predicts lattice distortion and defect formation energies and volumes with classical and machine-learning potentials, enabling high-dimensional uncertainty propagation without prohibitive overhead. We then show how the implicit derivative can be used to solve challenging inverse problems, minimizing an implicit loss to fine-tune potentials and stabilize solute-induced structural rearrangements at dislocations in tungsten.
2024
- Large-scale atomistic simulation of diffusion in refractory metals and alloysSergei Starikov, Petr Grigorev, Ralf Drautz, and Sergiy V DivinskiPhysical Review Materials 8 p. 43603 (Apr 2024)
The equilibrium vacancy concentration and atomic diffusion coefficients in dilute and complex refractory alloys have been calculated using various computational methods. The most productive technique has been large-scale atomistic simulation in the form of a numerical experiment in which a crystal with free surfaces was simulated for a relatively long time. This method is based on the concept that the free surface acts as a source of point defects and provides a natural way to achieve an equilibrium concentration of the defects within the bulk after an initial annealing stage. For complex concentrated alloys (CCAs), this numerical experiment offers the possibility to study diffusion processes where standard analytical approaches are difficult to apply due to the large variety of microscopic states. As the simulation results, we found that the transition from dilute alloys to CCA is accompanied by a significant increase in atomic diffusion rate due to both a substantial increase in the vacancy concentration and their mobility.
- matscipy: materials science at the atomic scale with PythonPetr Grigorev, Lucas Frérot, Fraser Birks, Adrien Gola, Jacek Golebiowski, Jan Grießer, Johannes L. Hörmann, Andreas Klemenz, Gianpietro Moras, Wolfram G. Nöhring, Jonas A. Oldenstaedt, Punit Patel, Thomas Reichenbach, Thomas Rocke, Lakshmi Shenoy, Michael Walter, Simon Wengert, Lei Zhang, James R. Kermode, and Lars PastewkaJournal of Open Source Software 9 p. 5668 (Jan 2024)
Behaviour of materials is governed by physical phenomena that occur at an extreme range of length and time scales. Computational modelling requires multiscale approaches. Simulation techniques operating on the atomic scale serve as a foundation for such approaches, providing necessary parameters for upper-scale models. The physical models employed for atomic simulations can vary from electronic structure calculations to empirical force fields. However, construction, manipulation and analysis of atomic systems are independent of the given physical model but dependent on the specific application. matscipy implements such tools for applications in materials science, including fracture, plasticity, tribology and electrochemistry.
- Angular-dependent interatomic potential for large-scale atomistic simulation of W-Mo-Nb ternary alloysSergei Starikov, Petr Grigorev, and Pär A.T. OlssonComputational Materials Science 233 p. 112734 (Jan 2024)
We present a new classical interatomic potential designed for simulation of the W-Mo-Nb system. The angular-dependent format of the potential allows for reproduction of many important properties of pure metals and complex concentrated alloys with good accuracy. Special attention during the development and validation of the potential was paid to the description of vacancies, screw dislocations and planar defects, as well as thermo-mechanical properties. Here, the applicability of the developed model is demonstrated by studying the temperature dependence of the elastic moduli and average atomic displacement in pure metals and concentrated alloys up to the melting point.
2023
- Nanoindentation of tungsten: From interatomic potentials to dislocation plasticity mechanismsF. J. Domínguez-Gutiérrez, P. Grigorev, A. Naghdi, J. Byggmästar, G. Y. Wei, T. D. Swinburne, S. Papanikolaou, and M. J. AlavaPhysical Review Materials 7 p. 043603 (Apr 2023)
In this study, we employed molecular dynamics simulations, both traditional and machine learned, to emulate spherical nanoindentation experiments of crystalline W matrices at different temperatures and loading rates using different approaches, such as EAM, EAM with Ziegler, Biersack, and Littmark corrections, modified EAM, analytic bond-order approach, and a recently developed machine-learned tabulated Gaussian approximation potential (tabGAP) framework for describing the W-W interaction and plastic deformation mechanisms. Results showed similarities between the recorded load-displacement curves and dislocation densities, for different interatomic potentials and crystal orientations at low and room temperature. However, we observe concrete differences in the early stages of elastic-to-plastic deformation transition, revealing different mechanisms for dislocation nucleation and dynamics during loading, especially at higher temperatures. This is attributed to the particular features of orientation dependence in crystal plasticity mechanisms and, characteristically, the stacking fault and dislocation glide energies information in the interatomic potentials, with tabGAP being the one with the most well-trained results compared to density functional theory calculations and experimental data.
-
 Calculation of dislocation binding to helium-vacancy defects in tungsten using hybrid ab initio-machine learning methodsPetr Grigorev, Alexandra M. Goryaeva, Mihai-Cosmin Marinica, James R. Kermode, and Thomas D. SwinburneActa Materialia 247 p. 118734 (Feb 2023)
Calculation of dislocation binding to helium-vacancy defects in tungsten using hybrid ab initio-machine learning methodsPetr Grigorev, Alexandra M. Goryaeva, Mihai-Cosmin Marinica, James R. Kermode, and Thomas D. SwinburneActa Materialia 247 p. 118734 (Feb 2023)Calculations of dislocation-defect interactions are essential to model metallic strength, but the required system sizes are at or beyond ab initio limits. Current estimates thus have extrapolation or finite size errors that are very challenging to quantify. Hybrid methods offer a solution, embedding small ab initio simulations in an empirical medium. However, current implementations can only match mild elastic deformations at the ab initio boundary. We describe a robust method to employ linear-in-descriptor machine learning potentials as a highly flexible embedding medium, precisely matching dislocation migration pathways whilst keeping at least the elastic properties constant. This advanced coupling allows dislocations to cross the ab initio boundary in fully three dimensional defect geometries. Investigating helium and vacancy segregation to edge and screw dislocations in tungsten, we find long-range relaxations qualitatively change impurity-induced core reconstructions compared to those in short periodic supercells, even when multiple helium atoms are present. We also show that helium-vacancy complexes, considered to be the dominant configuration at low temperatures, have only a very weak binding to screw dislocations. These results are discussed in the context of recent experimental and theoretical studies. More generally, our approach opens a vast range of mechanisms to ab initio investigation and provides new reference data to both validate and improve interatomic potentials.
2022
-
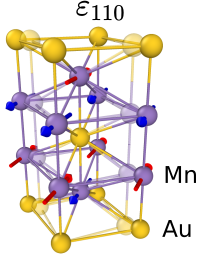 Optically Triggered Néel Vector Manipulation of a Metallic Antiferromagnet Mn2Au under StrainVladimir Grigorev, Mariia Filianina, Yaryna Lytvynenko, Sergei Sobolev, Amrit Raj Pokharel, Amon P. Lanz, Alexey Sapozhnik, Armin Kleibert, Stanislav Bodnar, Petr Grigorev, Yurii Skourski, Mathias Kläui, Hans-Joachim Elmers, Martin Jourdan, and Jure DemsarACS Nano 16 p. 20589-20597 (Nov 2022)
Optically Triggered Néel Vector Manipulation of a Metallic Antiferromagnet Mn2Au under StrainVladimir Grigorev, Mariia Filianina, Yaryna Lytvynenko, Sergei Sobolev, Amrit Raj Pokharel, Amon P. Lanz, Alexey Sapozhnik, Armin Kleibert, Stanislav Bodnar, Petr Grigorev, Yurii Skourski, Mathias Kläui, Hans-Joachim Elmers, Martin Jourdan, and Jure DemsarACS Nano 16 p. 20589-20597 (Nov 2022)The absence of stray fields, their insensitivity to external magnetic fields, and ultrafast dynamics make antiferromagnets promising candidates for active elements in spintronic devices. Here, we demonstrate manipulation of the Néel vector in the metallic collinear antiferromagnet Mn2Au by combining strain and femtosecond laser excitation. Applying tensile strain along either of the two in-plane easy axes and locally exciting the sample by a train of femtosecond pulses, we align the Néel vector along the direction controlled by the applied strain. The dependence on the laser fluence and strain suggests the alignment is a result of optically triggered depinning of 90° domain walls and their motion in the direction of the free energy gradient, governed by the magneto-elastic coupling. The resulting, switchable state is stable at room temperature and insensitive to magnetic fields. Such an approach may provide ways to realize robust high-density memory device with switching time scales in the picosecond range.
2021
- Efficient and transferable machine learning potentials for the simulation of crystal defects in bcc Fe and WAlexandra M. Goryaeva, Julien Dérès, Clovis Lapointe, Petr Grigorev, Thomas D. Swinburne, James R. Kermode, Lisa Ventelon, Jacopo Baima, and Mihai-Cosmin MarinicaPhysical Review Materials 5 p. 103803 (Oct 2021)
Data-driven, or machine learning (ML), approaches have become viable alternatives to semiempirical methods to construct interatomic potentials, due to their capacity to accurately interpolate and extrapolate from first-principles simulations if the training database and descriptor representation of atomic structures are carefully chosen. Here, we present highly accurate interatomic potentials suitable for the study of dislocations, point defects, and their clusters in bcc iron and tungsten, constructed using a linear or quadratic input-output mapping from descriptor space. The proposed quadratic formulation, called quadratic noise ML, differs from previous approaches, being strongly preconditioned by the linear solution. The developed potentials are compared to a wide range of existing ML and semiempirical potentials, and are shown to have sufficient accuracy to distinguish changes in the exchange-correlation functional or pseudopotential in the underlying referenc data, while retaining excellent transferability. The flexibility of the underlying approach is able to target properties almost unattainable by traditional methods, such as the negative divacancy binding energy in W or the shape and the magnitude of the Peierls barrier of the 1/2⟨111⟩ screw dislocation in both metals. We also show how the developed potentials can be used to target important observables that require large time-and-space scales unattainable with first-principles methods, though we emphasize the importance of thoughtful database design and degrees of nonlinearity of the descriptor space to achieve the appropriate passage of information to large-scale calculations. As a demonstration, we perform direct atomistic calculations of the relative stability of 1/2⟨111⟩ dislocations loops and three-dimensional C15 clusters in Fe and find the crossover between the formation energies of the two classes of interstitial defects occurs at around 40 self-interstitial atoms. We also compute the kink-pair formation energy of the 1/2⟨111⟩ screw dislocation in Fe and W, finding good agreement with density functional theory informed line tension models that indirectly measure those quantities. Finally, we exploit the excellent finite-temperature properties to compute vacancy formation free energies with full anharmonicity in thermal vibrations. The presented potentials thus open up many avenues for systematic investigation of free-energy landscape of defects with ab initioaccuracy.
2020
- Hybrid quantum/classical study of hydrogen-decorated screw dislocations in tungsten: Ultrafast pipe diffusion, core reconstruction, and effects on glide mechanismPetr Grigorev, Thomas D. Swinburne, and James R. KermodePhysical Review Materials 4 p. 023601 (Feb 2020)
The interaction of hydrogen (H) with dislocations in tungsten (W) must be understood in order to model the mechanical response of future plasma-facing materials for fusion applications. Here, hybrid quantum mechanics/molecular mechanics (QM/MM) simulations are employed to study the ⟨111⟩ screw dislocation glide in W in the presence of H, using the virtual work principle to obtain energy barriers for dislocation glide, H segregation, and pipe diffusion. We provide a convincing validation of the QM/MM approach against full DFT energy-based methods. This is possible because the compact core and relatively weak elastic fields of ⟨111⟩ screw dislocations allow them to be contained in periodic DFT supercells. We also show that H segregation stabilizes the split-core structure while leaving the Peierls barrier almost unchanged. Furthermore, we find an energy barrier of less than 0.05 eV for pipe diffusion of H along dislocation cores. Our quantum-accurate calculations provide important reference data for the construction of larger-scale material models.
2018
- Molecular dynamics simulation of hydrogen and helium trapping in tungstenPetr Grigorev, Aleksandr Zinovev, Dmitry Terentyev, Giovanni Bonny, Evgeny E. Zhurkin, Guido Van Oost, and Jean-Marie NoterdaemeJournal of Nuclear Materials 508 p. 451-458 (May 2018)
Tungsten has been chosen as the divertor armour material in ITER and is the main candidate material for plasma-facing components for future fusion reactors. Interaction of plasma components with the material leads to degradation of the performance and thus the lifetime of the in-vessel components. On top of that special attention is drawn to tritium retention in the reactors vessel from a safety point of view, since tritium is radioactive material. In order to gain better understanding of the mechanisms driving accumulation of plasma components in the material and subsequent degradation of the material, atomistic simulations are employed. The focus of this work is on so-called self trapping of H and He atoms or, in other words, Frenkel pair formation in bulk tungsten in the presence of H and He atoms. Two versions of a model embedded atom interatomic potential and a bond order potential were tested by comparing it with ab initio data regarding the binding properties of pure He and He-H-Vacancy clusters and energetics of Frenkel pair formation. As a result of Molecular Dynamics simulations at finite temperature, the values of critical H concentration needed for the generation of a Frenkel pair in the presence of He clusters were obtained. The results show that the critical H concentration decreases with the size of He cluster present in the simulation cell and thus, Frenkel pair formation by H is facilitated in the presence of He clusters in the material.
2017
- PhD ThesisAssessment of retention of plasma components in tungsten under high flux plasma exposure: multi-scale modelling approachPetr GrigorevPhD Thesis Ghent University, Faculty of Engineering and Architecture/Universidad Complutnese de Madrid, Facultad de Ciencias Fisicas (Apr 2017)
The results of my work described in this thesis are roughly divided in three parts. (1) Analysis of ab initio data and development of the model of dislocation mediated H retention in tungsten; (2) Development of an interatomic potential for W-H-He system and performing a large number of Molecular Dynamics (MD) simulations; (3) Implementation of the model in a Rate Theory simulation tool and its validation by comparison with experimental results; This pdf file also contains the pdf versions of the most of the articles listed below. Frankly speaking I am not allowed to share those here, but lets keep it between us.
- Trapping of hydrogen and helium at dislocations in tungsten: an ab-initio studyA. Bakaev, P. Grigorev, D. Terentyev, A. Bakaeva, E.E. Zhurkin, and Yu. A. MastrikovNuclear Fusion 57 p. 126040 (Oct 2017)
The interaction of H or He atoms with a core of edge and screw dislocations (SDs), with Burgers vector a0 <111>/2, is studied by means of ab initio calculations. The results show that the edge dislocations are stronger traps for H and He compared to the SDs, while the H/He affinity to both types of dislocation is significantly weaker than to a single vacancy. The lowest energy atomic configurations are rationalized on the basis of the charge density distribution and elasticity theory considerations. The results obtained contribute to the rationalization of the thermal desorption spectroscopy analysis by attributing certain peaks of the release of plasma components to the detrapping from dislocations. Complementary molecular statics (MS) calculations are performed to validate the accuracy of the recently developed W–H–He embedded atom method (EAM) and bond-order potentials. It is revealed that the EAM potential can reproduce correctly the magnitude of the interaction of H with both dislocations as compared to the ab initio results. All the potentials underestimate significantly the He-dislocation interaction and cannot describe correctly the lowest energy positions for H and He around the dislocation core. The reason for the discrepancy between ab initio and the MS results is rationalized by the analysis of the fully relaxed atomic configurations.
- Interaction of hydrogen and helium with nanometric dislocation loops in tungsten assessed by atomistic calculationsPetr Grigorev, Alexander Bakaev, Dmitry Terentyev, Guido Van Oost, Jean-Marie Noterdaeme, and Evgeny E. ZhurkinNuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 393 p. 164 - 168 (Feb 2017)
Abstract The interaction of H and He interstitial atoms with ½〈111〉 and 〈100〉 loops in tungsten (W) was studied by means of Molecular Static and Molecular Dynamics simulations. A recently developed interatomic potential was benchmarked using data for dislocation loops obtained earlier with two other W potentials available in literature. Molecular Static calculations demonstrated that ½〈111〉 loops feature a wide spectrum of the binding energy with a maximum value of 1.1 eV for H and 1.93 eV for He as compared to 0.89 eV and 1.56 eV for a straight ½〈111〉110 edge dislocation. For 〈100〉 loops, the values of the binding energy were found to be 1.63 eV and 2.87 eV for H and He, respectively. These results help to better understand the role played by dislocation loops in H/He retention in tungsten. Based on the obtained results, a contribution of the considered dislocation loops to the trapping and retention under plasma exposure is discussed.
2016
- Modelling deuterium release from tungsten after high flux high temperature deuterium plasma exposurePetr Grigorev, Dmitry Matveev, Anastasiia Bakaeva, Dmitry Terentyev, Evgeny E. Zhurkin, Guido Van Oost, and Jean-Marie NoterdaemeJournal of Nuclear Materials 481 p. 181-189 (Dec 2016)
Tungsten is a primary candidate for plasma facing materials for future fusion devices. An important safety concern in the design of plasma facing components is the retention of hydrogen isotopes. Available experimental data is vast and scattered, and a consistent physical model of retention of hydrogen isotopes in tungsten is still missing. In this work we propose a model of non-equilibrium hydrogen isotopes trapping under fusion relevant plasma exposure conditions. The model is coupled to a diffusion-trapping simulation tool and is used to interpret recent experiments involving high plasma flux exposures. From the computational analysis performed, it is concluded that high flux high temperature exposures (T = 1000 K, flux = 1024 D/m2/s and fluence of 1026 D/m2) result in generation of sub-surface damage and bulk diffusion, so that the retention is driven by both sub-surface plasma-induced defects (bubbles) and trapping at natural defects. On the basis of the non-equilibrium trapping model we have estimated the amount of H stored in the sub-surface region to be ∼10−5 at−1, while the bulk retention is about 4 × 10−7 at−1, calculated by assuming the sub-surface layer thickness of about 10 μm and adjusting the trap concentration to comply with the experimental results for the integral retention.
- Dislocation-mediated trapping of deuterium in tungsten under high-flux high-temperature exposuresA. Bakaeva, D. Terentyev, G. De Temmerman, K. Lambrinou, T.W. Morgan, A. Dubinko, P. Grigorev, K. Verbeken, and J.M. NoterdaemeJournal of Nuclear Materials 479 p. 307-315 (Oct 2016)
The effect of severe plastic deformation on the deuterium retention in tungsten exposed to high-flux low-energy plasma (flux ∼10^24 m−2 s−1, energy ∼50 eV and fluence up to 5 × 10^25 D/m2) was studied experimentally in a wide temperature range (460–1000 K) relevant for application in ITER. The desorption spectra in both reference and plastically-deformed samples were deconvoluted into three contributions associated with the detrapping from dislocations, deuterium-vacancy clusters and pores. As the exposure temperature increases, the positions of the release peaks in the plastically-deformed material remain in the same temperature range but the peak amplitudes are altered as compared to the reference material. The desorption peak attributed to the release from pores (i.e. cavities and bubbles) was suppressed in the plastically deformed samples for the low-temperature exposures, but became dominant for exposures above 700 K. The observed strong modulation of the deuterium storage in “shallow” and “deep” traps, as well as the reduction of the integral retention above 700 K, suggest that the dislocation network changes its role from “trapping sites” to “diffusion channels” above a certain temperature. The major experimental observations of the present work are in line with recent computational assessment based on atomistic and mean field theory calculations available in literature.
- Mobility of hydrogen-helium clusters in tungsten studied by molecular dynamicsPetr Grigorev, Dmitry Terentyev, Giovanni Bonny, Evgeny E. Zhurkin, Guido Oost, and Jean-Marie NoterdaemeJournal of Nuclear Materials 474 p. 143-149 (Jun 2016)
Tungsten is a primary candidate material for plasma facing components in fusion reactors. Interaction of plasma components with the material is unavoidable and will lead to degradation of the performance and the lifetime of the in-vessel components. In order to gain better understanding the mechanisms driving the material degradation at atomic level, atomistic simulations are employed. In this work we study migration, stability and self-trapping properties of pure helium and mixed helium-hydrogen clusters in tungsten by means of molecular dynamics simulations. We test two versions of an embedded atom model interatomic potential by comparing it with ab initio data regarding the binding properties of He clusters. By analysing the trajectories of the clusters during molecular dynamics simulations at finite temperatures we obtain the diffusion parameters. The results show that the diffusivity of mixed clusters is significantly lower, than that of pure helium clusters. The latter suggest that the formation of mixed clusters during mixed hydrogen helium plasma exposure will affect the helium diffusivity in the material.
- Numerical analysis of TDS spectra under high and low flux plasma exposure conditionsPetr Grigorev, Luxherta Buzi, Anastasia Bakaeva, Dmitry Terentyev, Gregory De Temmerman, Guido Van Oost, and Jean-Marie NoterdaemePhysica Scripta 2016 p. 014039 (Jan 2016)
A recently developed numerical model, based on the dislocation-driven nucleation of gas bubbles, is used to analyse experimental results on deuterium retention in tungsten under ITER relevant plasma exposure conditions. Focus is placed on understanding the relation between exposure temperature and flux on primary features of thermal desorption spectra: peak positions and intensities of the desorption flux. The model allows one to relate the peak positions with the size of plasma induced deuterium bubbles and envisage exposure conditions (temperature and flux) for their formation. Based on the performed analysis, dedicated experimental conditions to validate the model are proposed.
2015
- Interaction of hydrogen with dislocations in tungsten: An atomistic studyPetr Grigorev, Dmitry Terentyev, Giovanni Bonny, Evgeny E. Zhurkin, Guido Van Oost, and Jean-Marie NoterdaemeJournal of Nuclear Materials 465 p. 364-372 (Oct 2015)
The interaction of interstitial hydrogen with a dislocation and point defects in tungsten is studied by means of atomistic simulations. Two different types of interatomic potentials were tested by comparing their results with available ab initio data. The recently developed embedded atom method potential showed a better agreement with ab initio results than the bond order potential. Static calculations involving screw and edge dislocations showed that hydrogen is attracted to the dislocation core in both cases. It is also found that hydrogen atoms prefer to arrange themselves as elongated clusters on dislocation lines. Molecular dynamics simulations of hydrogen migration along the edge dislocation core confirmed the results of the static calculations and demonstrated a strong attraction to the dislocation core and one-dimensional migration along it.
- Nucleation and growth of hydrogen bubbles on dislocations in tungsten under high flux low energy plasma exposurePetr Grigorev, Dmitry Terentyev, Vladimir Dubinko, Giovanni Bonny, Guido Van Oost, Jean-Marie Noterdaeme, and Evgeny E. ZhurkinNuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 352 p. 96-99 (Jun 2015)
A new mechanism for the nucleation and growth of hydrogen (H) bubbles on dislocations under plasma exposure of tungsten was recently proposed on the basis of direct ab initio calculations. Density functional theory calculations demonstrated that H atoms are strongly bound to a screw dislocation core and exhibit fast one-dimensional migration along its line. Once the number of hydrogen atoms trapped on a dislocation segment exceeds eight, the emission of a jog occurs thereby converting a pure HN cluster into a HN+1-jog configuration. On the basis of these results a kinetic model was formulated to evaluate the conditions (i.e., range of temperature and flux exposure) for the transformation of pure H clusters into supercritical hydrogen–vacancy clusters attached to the dislocation line. In this work, a parametric study employing the kinetic nucleation model was performed to derive the hydrogen bubble formation energy function that offers the best agreement with available experimental results. The obtained results allow one to rationalize the depth and temperature dependence of the experimentally observed hydrogen deposition after high flux low energy plasma exposure for ITER relevant conditions.
2014
- On the binding of nanometric hydrogen–helium clusters in tungstenG. Bonny, P. Grigorev, and D. TerentyevJournal of Physics: Condensed Matter 26 p. 485001 (Oct 2014)
In this work we developed an embedded atom method potential for large scale atomistic simulations in the ternary tungsten–hydrogen–helium (W–H–He) system, focusing on applications in the fusion research domain. Following available ab initio data, the potential reproduces key interactions between H, He and point defects in W and utilizes the most recent potential for matrix W. The potential is applied to assess the thermal stability of various H–He complexes of sizes too large for ab initio techniques. The results show that the dissociation of H–He clusters stabilized by vacancies will occur primarily by emission of hydrogen atoms and then by break-up of V–He complexes, indicating that H–He interaction does influence the release of hydrogen.
- Dislocation mechanism of deuterium retention in tungsten under plasma implantationV. I. Dubinko, P. Grigorev, A. Bakaev, D. Terentyev, G. Oost, F. Gao, D. Van Neck, and E. E. ZhurkinJournal of Physics: Condensed Matter 26 p. 395001 (Aug 2014)
We have developed a new theoretical model for deuterium (D) retention in tungsten-based alloys on the basis of its being trapped at dislocations and transported to the surface via the dislocation network with parameters determined by ab initio calculations. The model is used to explain experimentally observed trends of D retention under sub-threshold implantation, which does not produce stable lattice defects to act as traps for D in conventional models. Saturation of D retention with implantation dose and effects due to alloying of tungsten with, e.g. tantalum, are evaluated, and comparison of the model predictions with experimental observations under high-flux plasma implantation conditions is presented.
- Many-body central force potentials for tungstenG. Bonny, D. Terentyev, A. Bakaev, P. Grigorev, and D Van NeckModelling and Simulation in Materials Science and Engineering 22 p. 053001 (Jun 2014)
Tungsten and tungsten-based alloys are the primary candidate materials for plasma facing components in fusion reactors. The exposure to high-energy radiation, however, severely degrades the performance and lifetime limits of the in-vessel components. In an effort to better understand the mechanisms driving the materials’ degradation at the atomic level, large-scale atomistic simulations are performed to complement experimental investigations. At the core of such simulations lies the interatomic potential, on which all subsequent results hinge. In this work we review 19 central force many-body potentials and benchmark their performance against experiments and density functional theory (DFT) calculations. As basic features we consider the relative lattice stability, elastic constants and point-defect properties. In addition, we also investigate extended lattice defects, namely: free surfaces, symmetric tilt grain boundaries, the 1/2〈111〉110 and 1/2〈111〉112 stacking fault energy profiles and the 1/2〈111〉 screw dislocation core. We also provide the Peierls stress for the 1/2〈111〉 edge and screw dislocations as well as the glide path of the latter at zero Kelvin. The presented results serve as an initial guide and reference list for both the modelling of atomically-driven phenomena in bcc tungsten, and the further development of its potentials.
- Dislocations mediate hydrogen retention in tungstenD. Terentyev, V. Dubinko, A. Bakaev, Y. Zayachuk, W. Van Renterghem, and P. GrigorevNuclear Fusion 54 p. 042004 (Mar 2014)
In this letter, a comprehensive mechanism for the nucleation and growth of bubbles on dislocations under plasma exposure of tungsten is proposed. The mechanism reconciles long-standing experimental observations of hydrogen isotopes retention, essentially defined by material microstructure, and so far not fully explained. Hence, this work provides an important link to unify material’s modelling with experimental assessment of W and W-based alloys as candidates for plasma facing components.
2013
- Transfer of molecular dynamics data to dislocation dynamics to assess dislocation–dislocation loop interaction in ironD. Terentyev, G. Monnet, and P. GrigorevScripta Materialia 69 p. 578-581 (Oct 2013)
We propose a computationally fast and physically justifiable method to treat dislocation loops as stochastic thermally activated finite-size obstacles in discrete dislocation dynamics simulations. The method was parameterized using molecular dynamics data for the interaction of dislocations with a0/2〈1 1 1〉 dislocation loops. As demonstration, the method is applied to rationalize experimental hardening of neutron-irradiated iron. The obtained results show good agreement with experimental data.
- Sputtering of Al nanoclusters by 1–13 keV monatomic or polyatomic ions studied by Molecular Dynamics simulationsEvgeny E. Zhurkin, and Petr Yu GrigorevNuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 303 p. 136-141 (May 2013)
Molecular Dynamics (MD) simulations were employed to study sputtering of freestanding and supported spherical Al nanoclusters with 2–10nm diameters under bombardment by Al1 and Al13 projectiles with energies of 1–13keV at normal and oblique incidence. Both monatomic and clustered yields of secondary emission are found to be larger than those for (111) flat surface of the bulk Al (at equal irradiation conditions). In some events, target nanocluster receives a backward momentum and therefore its major part (more than 1/2 of the mass) is ejected due to produced secondary emission mainly towards the substrate direction. This “recoil effect” is found more pronounced under the impact of cluster projectiles and its probability decreases with increase of the target cluster size. A restricted number of MD simulations were performed to verify whether this “recoil effect” is strong enough to desorb a 4nm Al nanocluster off an Al (111) substrate. Desorption was observed under oblique Al13 impact within the impact parameter range of 0.6–0.9.
- Simulation of the sputtering of Si nanoclusters with diameters of (2–8) nm under bombardment with monatomic and cluster ions using the method of classical molecular dynamicsP. Grigorev, and E. E. ZhurkinJournal of Surface Investigation. X-ray, Synchrotron and Neutron Techniques 7 p. 201–210 (Mar 2013)
The sputtering of Si nanoclusters with diameters of (2–8) nm under bombardment with Si 1 ions and Si 12 clusters with energies of 1 to 12 keV has been studied using the method of classical molecular dynamics. It is shown that the total sputtering yield is higher under the bombardment of nanoclusters with monatomic ions than in the case of the crystal plane surface mainly due to an increase in the contribution of the cluster component of the secondaryemission spectrum. In this case, in practice there is nonoccurrence of any correlation between the secondaryemission characteristics and the particleprojectile impact parameter. In the case of cluster bombardment, the cluster component of the spectrum of sputtered particles is dominant for large impact parameters (i.e., for glancing impact) and the sputtering yield and the probability of the ejection of large fragments (containing half of the initialnanocluster atoms or more) increase significantly (as compared with similar values obtained under bombardment with monatomic particles). It is established that the main mechanism for the emission of large fragments is the cocalled “recoil effect”, where the impinging particle projectile causes the secondary emission of nanocluster target atoms predominantly toward the substrate; as a result, the clustertarget acquires momentum in the opposite direction. It is shown that this “recoil effect” can lead to the desorption of the nanocluster, deposited onto a plane Si(111) substrate.